
Inherited Cardiomyopathies and Genetics
Dr Tiziana Felice
ABSTRACT
Cardiomyopathies are a clinically heterogeneous group of cardiac muscle disorders. They are defined by the presence of abnormal myocardial structure in the absence of ischaemic and valvular heart disease, hypertension and congenital heart disease. There are five main types of inherited cardiomyopathies: hypertrophic, dilated, arrhythmogenic right ventricular cardiomyopathy, restrictive and left ventricular non-compaction cardiomyopathy. For most cardiomyopathies autosomal dominant transmission is the commonest mode of inheritance except for those caused by metabolic disorders. Cardiomyopathies are associated with the early development of heart failure and an increased risk of sudden cardiac death often claiming the lives of young patients. Advances in molecular genetics have allowed us to better understand these myocardial diseases allowing for better clinical diagnosis, management and familial screening. This review will focus on the genetics in HCM and DCM.
Key words: Hypertrophic cardiomyopathy, Dilated cardiomyopathy, Heart failure, Sarcomere, Genetics
INTRODUCTION
Cardiomyopathies are disorders causing abnormalities primarily in the structure and function of the heart in the absence of coronary artery disease, valvular heart disease and hypertension. These disorders are commonly grouped into morphological subtypes that include hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and left ventricular non-compaction cardiomyopathy (LVNC).1,2 Early clinical investigations showed familial transmission in individuals with cardiomyopathies suggesting a critical role for genetics. Research advances over the past thirty years confirmed this hypothesis and today many cardiomyopathies are recognized as monogenic disorders. Most cardiomyopathies are often inherited in an autosomal dominant manner. This literature review will focus on the genetics of HCM and DCM.
GENETICS IN HYPERTROPHIC CARDIOMYOPATHY
HCM is defined by the presence of increased left ventricular (LV) wall thickness that is not explained by abnormal loading conditions, such as hypertension.3 An index case is diagnosed with HCM if the LV wall thickness is ≥ 15mm in one or more myocardial segments, whilst first-degree relatives are diagnosed with the condition if the LV wall thickness is ≥ 13mm.3 The above definitions are in accordance with the 2014 European Society of Cardiology guidelines on the diagnosis and management of hypertrophic cardiomyopathy.
HCM is a monogenic disease caused by variants in genes that encode the protein components of the cardiac sarcomere. HCM is the commonest inherited cardiac disease with an incidence in the population of 1 in 500.4 Therefore in Malta it is estimated that approximately 900 individuals will be affected with this condition. HCM is transmitted in an autosomal dominant manner and therefore first-degree relatives of affected individuals have a 50% chance of developing the disease. It is the commonest cause of sudden cardiac death (SCD) in young athletes, and may also progress to heart failure.4 Penetrance is often incomplete and individuals express the clinical phenotype later on in life, in adolescence or adulthood.5 Identical HCM variants within the same family may result in varying degrees of disease phenotype and severity. The morphology of cardiac muscle affected with HCM is characterized by cardiac myocyte hypertrophy, myocyte disarray and increased myocardial fibrosis. The cardiac myocytes have distorted nuclei and disorganised myofibrils. These changes alone may lead to decreased cardiac relaxation, arrhythmias and progression to heart failure.4 Figure 1 shows the different types of HCM depending on the location and morphology of the hypertrophied segment.
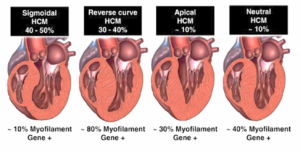
Figure 1. Septal Morphologies in HCM. Shown are the most common septal morphologies in HCM. The distribution of septal morphologies among a large cohort of patients with HCM is shown along the top while the yield of genetic testing for each morphological subgroup is shown along the bottom of the figure. Bos JM, et al. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J AM Coll Cardiol 2009;54:201-211.
Individuals affected with HCM may have a variety of symptoms. They often complain of palpitations, chest pain or shortness of breath. Unheralded syncope may occur and this may precede sudden cardiac arrest as it is often caused by a malignant ventricular arrhythmia. Patients are more symptomatic in the presence of left ventricular out flow tract (LVOT) obstruction where they may develop chest pain, dyspnoea or dizziness on walking uphill or upstairs which are worse after eating a heavy meal. The management of patients with HCM involves treating symptoms and protecting patients from SCD. Drugs worsening the gradient across the LVOT should be withheld, such as ACE-inhibitors, nitrates, dihydropyridine calcium channel blockers and digoxin. LVOT obstruction is treated with beta-blockers, non-dihydropyridine calcium channel blockers or disopyramide. If symptoms persist surgical septal myectomy or alcohol septal ablation may be considered. A patient’s risk for SCD is assessed using the European Society of Cardiology’s HCM-SCD risk score and if the calculated risk is ≥ 6%, an implantable cardiac defibrillator will be inserted.3 This risk score takes into consideration the maximal wall thickness, the left atrial diameter, the LVOT gradient, any family history of SCD in a first-degree relative age <45 years, the presence of non-sustained ventricular tachycardia on holter, and syncope.
The first identified HCM–causing mutation was discovered in 1990. Since then several studies have demonstrated disease-causing variants in eight genes: β-myosin heavy chain (MYH7),6 α-tropomyosin (TPM1), cardiac troponin T (TNNT2),7 cardiac myosin binding protein-C (MYBPC3),8 myosin regularity light chain (MYL2), myosin essential light chain (MYL3),9 cardiac troponin I (TNNI3)10 and cardiac α-actin (ACTC1).11 Most of these genes encode proteins of the myofilaments or Z-disc of the sarcomere as shown in Figure 2. More than 1,400 mutations have been described in association with HCM. Pathogenic variants affecting the MYBPC3 and MYH7 proteins are responsible for up to 50% of all clinically recognized cases of HCM. A disease–causing pathogenic variant is found in approximately 30-60% of HCM probands.
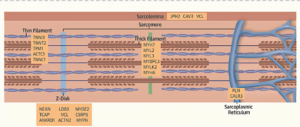
Figure 2. A Schematic of Definitive and Posited HCM genes with the Subcellular Localisation of the Encoded Proteins. Burke MA, et al. Clinical and Mechanistic Insights into the genetics of Cardiomyopathy. J Am Coll Cardiol 2018;68:2871-2886.
These genetic variants affect the biochemical and mechanical function in several ways. The myosin head region is the domain that generates force, hydrolyzes adenosine triphosphate and interacts with regulatory light chain, troponin T, and actin. Mutations in this domain exhibit biophysical properties that enhance contraction, but impair relaxation. Mutations in MYBPC3 modulate contractile performance through interaction with myosin and titin. HCM mutations increase the energetic cost of contraction and also cause increase in calcium release causing diastolic dysfunction.12
The large numbers of HCM-causing genetic variants in different sarcomeric genes together with genetic, epigenetic and environmental modifiers explains why the genotype alone cannot predict patient specific clinical manifestations of HCM. There are several MYH7 variants that cause significant cardiac remodeling with an increased risk of SCD and an increased risk for developing end-stage heart failure.13 Some TTN2 variants are associated with less hypertrophy but a higher risk in SCD.7 Having more than one pathogenic variant results in a worse phenotype and prognosis.13
GENETICS IN DILATED CARDIOMYOPATHY
DCM is characterized by the enlargement of the left ventricular chamber accompanied with systolic dysfunction.1 Histologically myocyte enlargement, cellular apoptosis and myocardial fibrosis are present, causing arrhythmias and heart failure. This cardiomyopathy is the commonest referral for heart transplantation. Around 50% of non-ischaemic DCM have no obvious aetiology, and in both familial and sporadic cases, genetic causes are increasingly identified.14 Early population echocardiography studies estimated the prevalence of unexplained DCM as 1:2,500.15 but recently epidemiological and heart failure data suggest a much higher prevalence of DCM as high as 1:250.16 This matches the prevalence of likely pathogenic variants identified by next-generation sequencing.17 Therefore there should be approximately 180-1,800 individuals affected with this condition in the Maltese population. Figure 3 shows cardiac MRI images comparing a healthy heart to one affected with DCM.
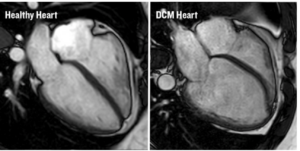
Figure 3. CMR Images of a Healthy Heart and a DCM Heart. https://www.bhf.org.uk/what-we-do/news-from-the-bhf/news-archive/2018/march/largest-ever-study-of-killer-heart-condition.
Patients with DCM may be asymptomatic at first but they often present with acute shortness of breath, palpitations and lower limb oedema. Again, unheralded syncope may occur in this group of patients secondary to ventricular arrhythmias. The treatment in DCM is to prevent further negative remodeling of the myocardium and protect the individual from SCD. Fluid overload is treated with loop diuretics but aggressive medical treatment is commenced to improve the left ventricular function. This medication includes ACE-inhibitors, carvedilol and spironolactone. Novel medications such as angiotensin receptor-neprilysin inhibitors and dapagliflozin have been found to further improve the myocardial strength and together with the previously mentioned medications, also reduce hospitalization and mortality. If patients remain symptomatic at NYHA II or more with an EF of ≤35% despite optimal medical therapy, an implantable cardiac defibrillator or cardiac resynchronisation therapy (depending on the width of the QRS on ECG) should be considered.
In DCM, most genetic variants are transmitted as dominant traits but a few exhibit recessive, X-linked and matrilinear inheritance. DCM-causing genetic variants exhibit incomplete and age-dependent penetrance with the phenotype expression being delayed until the fifth and sixth decade. The substantial genetic heterogeneity of DCM has made comprehensive genetic testing technically difficult and costly. Next-generation sequencing has allowed us to fully interrogate all DCM genes and this should substantially increase mutation detection in both familial and sporadic cases. DCM-causing genes encode for a diverse group of proteins that are important in: 1) generating and transmitting force, 2) sarcomere integrity, 3) cytoskeletal and nuclear architecture, 4) electrolyte homeostasis, as well as 5) mitochondrial function and transcription.
Pathogenic variants in the titin (TTN) gene, which encodes for the titin protein in the sarcomere are the commonest cause of DCM accounting for 15 to 20% of cases.18 Mutations that truncate the titin protein, such as non-sense, frameshift, or splice-site variants co-segregate in familial DCM and display nearly complete penetrance after 40 years of age. Phospholamban variants cause DCM by altering calcium homeostasis as it regulates calcium uptake by the sarcoplasmic/endoplasmic reticulum. The Arg9Cys mutation in PLN is particularly severe causing progressive heart failure requiring early heart transplantation.19 Some mutations in SCN5A have also been implicated in DCM. This gene encodes for the sarcolemmal transmembrane cardiac voltage–gated sodium channel, that functions in developing cardiac action potentials. SCN5A mutations cause a high burden of arrhythmias and are responsible for causing Brugada syndrome, idiopathic ventricular fibrillation and familial atrial fibrillation.20
Multiple DCM genes cause both heart and skeletal muscle phenotypes, including LMNA, which encodes for the protein lamin A/C, a protein expressed in the inner nuclear membrane that plays a role in the maintenance of the proper nuclear structure. LMNA mutations occur in approximately 6% of DCM cases and also cause conduction disease. In fact, conduction disease and atrial fibrillation often precede DCM that inevitably leads to heart failure.21 LMNA mutations are highly predictive of progressive conduction disease and risk of SCD. Early assessment for prophylactic implantable cardioverter–defibrillator (ICD) placement to treat malignant ventricular arrhythmias should be considered.22 Figure 4 is a schematic diagram showing the various DCM-causing genes and the location of their encoded protein within the myocyte.
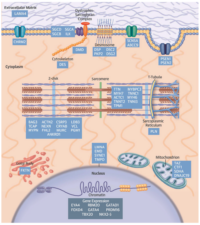
Figure 4. Schematic of Definitive and Posited DCM Genes with the Subcellular Localization of the Encoded Proteins. Pathogenic genes encode proteins that participate in many diverse biological processes of cardiomyocytes. Burke MA, et al. Clinical and Mechanistic Insights into the Genetics of Cardiomyopathy. J Am Coll Cardiol 2016;68(25): 2871-2886.
GENETIC TESTING IN CLINICAL PRACTICE
Knowledge of the genetic cause of a cardiomyopathy in patients can improve clinical management and provides diagnostic certainty. It may also help to guide the use of emerging therapies that target the biophysical consequences associated with the mutations.23 Genetic diagnosis enables cost-effective screening of first-degree relatives, eliminating health care expenditures.24 Cardiomyopathy gene panels continue to evolve thanks to next-generation sequencing and include comprehensive analysis of all genes implicated in HCM, DCM, ARVC and LVNC. If a disease-causing variant is not found, one may also use whole exome and whole genome sequencing since now the cost of genetics testing has become more affordable. Genetic testing may yield five distinct results: 1) identification of a definitely pathogenic variant. This outcome confirms the diagnosis, establishes the aetiology and identifies a target for familial screening, 2) identification of a probable pathogenic mutation, this potentially supports the clinical diagnosis but additional evidence such as familial co-segregation would be necessary to establish causality, 3) identification of a variant of unknown significance. This does not distinguish whether the variant is disease-causing or whether it represents a rare polymorphism unrelated to the disease and 4) identification of benign variants which are not disease-causing and occur in the general population.25
A cardiovascular genetic service has been setup at Mater Dei Hospital in 2020 in a joint effort between the cardiology and genetics departments. Thanks to this service cardiomyopathy patients who are seen at the inherited cardiomyopathy clinic may have genetic studies performed. When a definite pathogenic variant is found in the proband, targeted cascade genetic screening may be done on the first-degree relatives. In this way first-degree relatives who are genotype negative may be discharged from the clinical screening program. On the other hand regular clinical screening will continue on the genotype -positive individuals as seen in Figure 5.
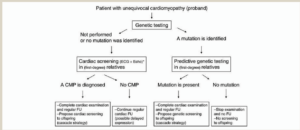
Figure 5. Organization of Family Screening. Charron P, et al. Genetic counseling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial diseases. EHJ 2010;31:2715-2728.
Unfortunately there are times when cardiomyopathies present for the first time as SCD. In this situation genetics on the tissues may help us find the genetic variant responsible and hopefully protect the rest of the family. A family history of sudden death at a young age should alert one to the possibility of inherited cardiac disorders as a cause and referral for assessment of the relatives by a cardiologist is recommended.
CONCLUSION
Cardiomyopathies are a group of diverse disorders affecting the heart muscle with an increasing morbidity and mortality often affecting the young. Early diagnosis of these conditions allows for earlier and more aggressive management of this cohort of patients. Genetic studies have allowed us to improve the management of our cardiomyopathy patients as well as their relatives.
REFERENCES
- Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation 2006;113:1807-16.
- Elliott P, Anderson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working group in Myocardial and Pericardial diseases. European Heart J 2008;29:270-6.
- Elliot PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on the diagnosis and management of hypertrophic cardiomyopathy. European Heart Journal 2014;39:2733-2779.
- Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Circulation 1995;92:785-9.
- Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics. J Am Coll Cardiol 2018;68:2871-2886.
- Geisterfer-Lowrance AAT, Kass S, Tnigawa G, et al. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell 1990;62:999-1006.
- Thierfelder L, Watkins H, MacRae C, et al. α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 1994;77:701-12.
- 8.Watkins H, Conner D, Thierfelder L, et al. Mutations in the cardiac myosin binding protein- C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 1995;11:434-7.
- Poetter K, Jiang H, Hassanzadeh S, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 1996;13:63-9.
- Kimura A, Harada H, Park JE, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997;16(4):379-82.
- Mogensen J, Klausen IC, Pedersen AK, et al. α-Cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest 1999;103:39-43.
- Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J 2014;106:1236-49.
- Tesson F, Richard P, Charron P, et al. Genotype–phenotype analysis in four families with mutations in β-myosin heavy chain gene responsible for familial hypertrophic cardiomyopathy. Hum Mutat 1998;12:385-92.
- asper EK, Agema WR, Hutchins GM, et al. The causes of dilated cardiomyopathy: a clinicopathologic review of 673 consecutive patients. J Am Coll cardiol 1994;23:586-90.
- Codd MB, Sugrue DD, Gersh BJ, et al. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation 1989;80:564-72.
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531-47.
- Golbus JR, Puckelwartz MJ, Fahrenbach JP, et al. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet 2012;5:391-9.
- Herman DS, Lam L, Taylor MRG, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619-28.
- Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban . Science 2003;299:1410-3.
- McNair WP, Ku L, Taylor MRG, et al. Familial Cardiomyopathy Registry Research Group. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004;110:2163-7.
- Taylor MRG, Fain PR, Sinagra G, et al. Familial Dilated Cardiomyopathy Registry Research Group. Natural History of dilated cardiomyopathy due to LaminA/C gene mutations. J AM Coll Cardiol 2003;41:771-80.
- Van Rijingen IAW, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European cohort study. J Am Coll Cardiol 2008;52:1250-60.
- Green EM, Wakimoto H, Anderson RL, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016;351:617-21.
- Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy; expanded panels offer limited additional sensitivity. Genet Med 2015;17:880-8.
- Richard S, Aziz N, Bale S, et al. ACMG Laboratory Quality Assurance Commitee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24.