
Diagnosis of BETA Thalassaemia
Prof. Joseph Borg and Dr Laura Grech
HAEMOGLOBIN Haemoglobin (Hb) is chemically best considered as a duplex of globin heterodimers. The main adult haemoglobin (HbA) is composed of two alpha (α) and two beta (β) chains assembled in two αβ dimers (α1 β1 and α2 β2 ), while foetal haemoglobin (HbF) is composed of two alpha-gamma (αγ) dimers.1 Their main function is to transport oxygen from the lungs to tissues, but it also specifically interacts with three other gases, carbon dioxide, carbon monoxide and nitric oxide.
The α chains are located on the short arm of chromosome 11 while the β chains are located on the short arm of chromosome 16. In the first stages of human development, embryonic haemoglobin consisting of two heterodimers of ε and ζ-globin chains is expressed in red blood cell progenitors (figure 1). At 12 weeks post-conception, as the site of erythropoiesis changes from the yolk sac to foetal liver the first switch in globin composition occurs. The embryonic haemoglobin is replaced by HbF consisting of α- and γ- globin chains.2 Around birth as erythropoiesis starts taking place in the bone marrow and spleen, the second globin switch occurs. This results in the decline of HbF synthesis together with increased synthesis of adult haemoglobin composed of HbA (α2 β2 ) with a minor HbA2 (α2 δ2 ).3,4 Residual amounts of HbF continue to be synthesized throughout adult life and expressed by F-erythrocytes.5 In most adults, F-erythrocytes contain less than 2% of total Hb, although there is considerable variation6 (figure 1).
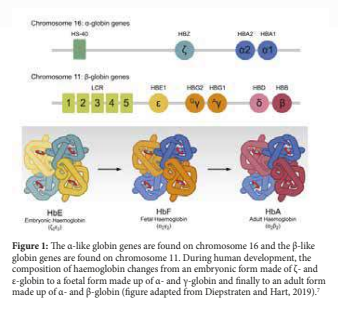
HAEMOGLOBINOPATHIES
Mutations in the beta globin gene can give rise to haemoglobinopathies. Haemoglobinopathies are the commonest monogenic disorders, affecting about 7% of the world’s populations.8 The haemoglobinopathies can be classified as (i) the structural variants such as Sickle cell disease (HbS), haemoglobin C (HbC) and Haemoglobin E (HbE), (ii) thalassaemia which can be further sub-classified into α, β, δβ and εγδβ thalassaemia depending on the particular globin chain or chains that is/are ineffective synthesized, and (iii) Hereditary Persistence of Foetal Haemoglobin (HPFH) in which there is a defect in the normal switch from foetal to adult haemoglobin.9
BETA-THALASSAEMIA
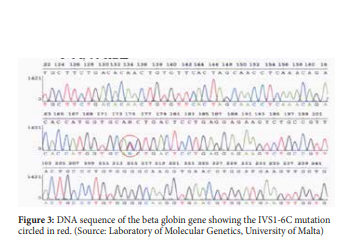
β-thalassaemia is most commonly present in persons of Mediterranean, African and Southeast Asian descent. β-thalassaemia can be divided into three main forms which are the β-thalassaemia major also known as Cooley anaemia, β-thalassaemia Intermedia and β-thalassaemia minor also called β-thalassaemia trait or β-thalassaemia carrier. It is the result of deficient or absent synthesis of beta globin chains which are controlled by one gene on each chromosome 11. The degree of globin chain reduction is determined by the nature of the mutation in the β-globin gene.10 In the Maltese population, the most common mutations are the IVS1-6C, IVS1-110A, IVSII-1A and COD39T.11 The β+ IVS1-6C mutation is the most predominant β-thalassaemia mutation in the Maltese population.12
DIAGNOSIS OF BETA-THALASSAEMIA TRAIT
The diagnosis of β-thalassaemia begins with suspicion of the disease in anaemic patients based on phenotype, family history and relevant laboratory screening. The detection and characterisation involve a 3 tier-work up: (1) Complete blood count and picture, (ii) Special haematological tests, and (iii) DNA testing.
- COMPLETE BLOOD COUNT AND PICTURE For screening of β-thalassaemia, the mean corpuscular volume (MCV) of less than 80fL and/or mean corpuscular haemoglobin (MCH) value of less than 26pg are usually used as cut-off levels for a positive screening result. The mean corpuscular haemoglobin concentration (MCHC) is usually normal. The advantages of thalassaemia screening by MCV and MCH are the rapid, cost effective, reproducibility and accurate analysis.13 One has to keep in mind that several conditions such as iron deficiency anaemia and anaemia of inflammation can also results in low MCV and MCH. Also, normal ranges of MCV vary by age in infants and young children. On the other hand, the interaction of β-thalassaemia trait with α-thalassaemia trait alone or together with glucose-6-phosphate dehydrogenase deficiency may lead to normal MCV and gives a false-negative result for thalassaemia screening.13 The haemoglobin concentration can be normal or slightly reduced in β-thalassaemia carriers while the red blood cell (RBC) count can be elevated. The red cell distribution width (RDW) is a measure of the degree of variations in red cell size, and an increase in RDW can be noticed in β-thalassaemia carriers.14 In β-thalassaemia carriers, the blood film varies from almost normal, with only mild microcytosis, to markedly abnormal. In addition to microcytosis abnormal features can include anisocytosis, hypochromia and poikilocytosis. Prominent basophilic stippling and target cells can be seen in individuals with a more severe phenotype.
Screening of thalassemia carriers by using RBC indices alone in a population with a high prevalence of β-thalassemia is not sufficient and other techniques such as haemoglobin analysis and DNA testing are required.
- HAEMOGLOBIN ANALYSIS
Haemoglobin analysis is an important laboratory evaluation to provide a presumptive identification and diagnosis of β-thalassaemia. There are several platforms for haemoglobin analysis including haemoglobin electrophoresis using cellulose acetate membrane, acid agarose or citrate agar gel, isoelectric focusing (IEF), high performance liquid chromatography (HPLC) and capillary electrophoresis. Each method uses different principles to separate different species of haemoglobin molecules.15 In Malta for the diagnosis of the β-thalassaemia trait we use the IEF together with HPLC.
Isoelectric focusing
IEF uses a polyacrylamide or agarose gel containing low molecular weight molecules. These molecules allow the establishment of a pH gradient; for separation of haemoglobins the pH ranges from 6-8. When an electric current is applied to the gel, the amphoteric molecules migrate through the gel to their isoelectric points along the gel. In doing so they form a stable pH gradient. Haemoglobin variants also exhibit this characteristic. They migrate through the gel until they reach the area in which their individual isoelectric point is equal to the corresponding pH on the gel. When these occur the charges on the variants are 0 and therefore migration ceases. The electric field counteracts diffusion and the haemoglobin variant forms a discrete thin band. The different Hb variants have different isoelectric point and thus they separate out on the gel to form bands at specific positions. The bands are then stained.15 In β-thalassaemia carriers the HbA is reduced while HbF and HbA2 are slightly increased
High performance liquid chromatography
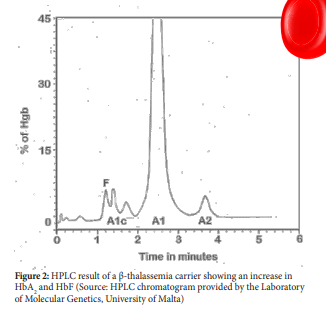
HPLC technique is a method used to separate compounds or molecules on the basis of their chemical characteristics. Separation of Haemoglobin is carried out using the VARIANT β-thalassaemia Short Program which utilizes the principles of cation-exchange HPLC. The HPLC rapidly and accurately measures HbA2 which often provides a diagnostic clue to the presence of β-thalassaemia trait especially when this is found with hypochromic, microcytic erythrocytes.
The increase in HbA2 in β-thalassaemia carriers is a result of both transcriptional and post-transcriptional effects and an increase of more than 3.2% can indicate the presence of β-thalassaemia trait (figure 2). In β-thalassaemia carriers the HbA2 levels vary according to the thalassaemia mutation. Small deletions of the 5’ portion of the beta globin gene are associated with the highest HbA2 levels. In fact, an HbA2 of 7-9% is seen in heterozygotes that have deletions that involves the remove of the beta globin promoter.16,17
From studies it is now clear that increases in HbA2 may not be a constant accompaniment of β-thalassaemia trait. β-thalassaemia trait patients who also have iron deficiency have slightly reduced but still elevated HbA2 but β-thalassaemia trait individuals with severe iron deficiency can have reduced HbA2 levels (less than 3.0%).17 On the other hand, individuals with variants in KLF1 can also have an increase in HbA2 levels and if their HbA2 levels fall between 3.3% and 3.9% it is important that these subjects are not classified as β-thalassaemia trait.18,19
- DNA ANALYSIS
β-thalassaemia is mainly caused by mutations in globin gene and therefore the most definitive diagnosis for β-thalassaemia is the molecular analysis of the beta globin sequence (figure 3). Many different molecular techniques are used to detect beta globin mutations. These molecular techniques can be grouped by the mutation type they target: (i) detection methods for structural variations such as gene deletion and duplication, and (ii) detection methods for sequence variations such as nucleotide substitution, insertion or short insertion/deletions.20
CONCLUSION
β-thalassaemia mutations are inherited in an autosomal recessive manner. Two β-thalassaemia heterozygote parents have a 25% chance of having a β-thalassaemia major child and thus, proper diagnosis of β-thalassaemia trait is important. Diagnosis of β-thalassaemia requires a comprehensive evaluation combining red blood cell phenotypes, haemoglobin profiles and DNA analysis
REFERENCES
- Perutz MF. Stockholm: Les Prix Nobel; 1963. X-Ray Analysis of Haemoglobin.
- Dover GJ, Boyer SH. Quantitation of hemoglobins within individual red cells: Asynchronous biosynthesis of fetal and adult hemoglobin during erythroid maturation in normal subjects. Blood 1980; 56(6):1082-1091.
- Brinkman R, Jonxis J H. The occurrence of several kinds of haemoglobin in human blood. J Physiol 1935; 85(2):117-127
- Weinberg R S, Goldberg JD, Schofield JM et al. Switch from fetal to adult hemoglobin is associated with a change in progenitor cell population. J Clin Invest 1983; 71(4):785-794.
- Hosoi T. Studies on hemoglobin F within single erythrocyte by fluorescent antibody technique. Exp Cell Res1965; 37:680-683.
- Thein SL, Menzel S, Lathrop M. et al. Control of fetal hemoglobin: New insights emerging from genomics and clinical implications. Hum Mol Genet 2009;18(R2): R216-23.
- Diepstraten ST and Hart AH. Modelling human haemoglobin switching. Blood Reviews 2019; 33:11-23
- Manca L, Masala B. Disorders of the synthesis of human fetal hemoglobin. IUBMB Life 2008; 60(2):94-111.
- Weatherall DJ. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat Rev Genet 2001; 2(4):245-255.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis 2010; 21:5-11.
- Scerri CA, Abela W, Galdies R et al. The beta + IVS, I-NT no. 6 (T –> C) thalassaemia in heterozygotes with an associated Hb Valletta or Hb S heterozygosity in homozygotes from Malta. Br J Haematol 1993;83(4):669-71.
- Felice AE, Abela W, Bartolo C etal. Quantification of in vivo expression of the b-IVS-I-NT no. 6 thalassaemia mutation. (Abstract) Proceedings, Fourth lnternational Conference on Thalassaemia and the Haemoglobinopathies, 1991, Nice, France, p.101.
- Viprakasit V, Limwongse C, Sukpanichnant S, Ruangvutilert P, Kanjanakorn C, Glomglao W, Sirikong M, Utto W, Tanphaichitr VS. Problems in determining thalassemia carrier status in a program for prevention and control of severe thalassemia syndromes: a lesson from Thailand. Clin Chem Lab Med 2013;51(8):1605-14.
- Clarke GM, Higgins TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem 2000;46(8 Pt 2):1284-90.
- Bain BJ, Wild BJ, Stephens AD, Phelan L. Variants Haemoglobins: A guide to identification. Oxford: Wiley-Blackwell; 2010.
- Codrington JF, Li HW, Kutlar F et al. Observations on the levels of hb A2 in patients with different beta-thalassemia mutations and a delta chain variant. Blood 1990;76(6): 1246-1249.
- Verhovsek M, So CC, O’Shea T, et al. Is HbA2 level a reliable diagnostic measurement for β-thalassemia trait in people with iron deficiency? Am J Hematol 2012;87(1):114-6.
- Grech L. Genetic heterogeneity of KLF1 deficiency and the pleiotropy of haemoglobin phenotypes (Doctoral dissertation), University of Malta, Malta. 2019.
- Perseu L, Satta S, Moi P, et al. KLF1 gene mutations cause borderline HbA(2). Blood 2011; 118(16):4454-4458.
- Viprakasit V, Ekwattanakit S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol Oncol Clin North Am 2018;32(2):193-211.