
Coeliac Disease – The Wandering Iceberg
by Christian A Scerri MD PhD (Molecular Genetics)
Consultant Geneticist
Mater Dei Hospital
Coeliac disease (CD), also known as coeliac sprue, non-tropical sprue, and gluten-sensitive enteropathy, is an inflammatory disease of the upper small intestine that results from gluten ingestion in genetically susceptible individuals1. The characteristic mucosal inflammation, villous atrophy and crypt hyperplasia that occur upon exposure to gluten leads to malabsorption of several important nutrients. CD was first formally described in 1888, by Samuel Gee in the St Bartholomew’s Hospital reports2. The central role of gluten in the pathogenesis of CD was observed by Dicke in 19503. He noted that persons previously diagnosed with CD improved during World War II when grain products were in short supply whilst when grains became more plentiful after the war, the incidence of coeliac disease returned to its pre-war levels. CD is associated with dermatitis herpetiformis4 a typical skin rash that similarly to CD responds to withdrawal of gluten. Untreated CD is associated with multiple, short and long-term complications including nutritional derangements, anaemia, reduced bone density, as well as intestinal lymphoma.
Though its triggering factor (surgery, pregnancy, childbirth, viral infections and stress have all been implicated at some time or other) and its polygenic basis is still mostly unknown, CD is unique amongst the inflammatory diseases in that the causative agent is known, i.e. gluten. This fact projects CD as a possible model for the study of inflammatory disease.
In the 1980’s, Marsh demonstrated that the immune system played an important role in causing intestinal injury in coeliac disease5. It has been shown that CD is the result of the activation of both a cell-mediated (T-cell) and humoral (B-cell) immune response upon exposure to the glutens (prolamins and glutenins) of wheat, barley, rye, and oats, in a genetically susceptible person6.
CD was thought to be uncommon but in the last two decades, it has been realised that the condition is relatively common, affecting 1 in every 100 persons7-10 to 1 in 50 persons11. Traditionally CD was regarded as a childhood disease but it is now clear that CD can occur at any age, with the fifth decade being the peak incidence in adults. Females are more commonly affected than males with a female to male ratio of around 3 to 1. The increased incidence of the condition can be partly explained through the greater awareness of its presentation and the availability of accurate serologic tests, though an actual increase as a result of as yet unknown triggering factors cannot be ruled out. The serological tests that helped to put CD in the forefront are the anti-gliadin antibodies (IgA and IgG), anti-endomyseal antibodies and recently the identification of anti-Transglutaminase antibody. The latter test has a very high sensitivity and specificity (99% and 98% respectively) making it ideal as an initial screening test.
Genetic predisposition, as indicated by the high concordance between monozygotic twins and the high prevalence amongst family members of affected individuals, plays a major role in CD. In the last decade or so considerable progress has been made in identifying genes that are responsible for CD predisposition. HLA Class II genes have been positively identified as a predisposing genetic factor for CD. The condition is strongly associated with the specific HLA class II genes known as HLA-DQ2 and HLA-DQ8 located on chromosome 6p21. HLA-DQ2 is found in the majority of CD patients (95%) whilst the remaining are usually HLA-DQ8 positive. As the HLA-DQ2 allele is very common in the population (around 30% of Caucasian individuals), it was clear that though the HLA type background is necessary for CD, it is not sufficient for CD to develop. The HLA locus has been assigned as the CELIAC1 locus. HLA testing, specifically for the DQ2 and DQ8 alleles, is useful as an exclusive test, i.e. those individuals that are negative for the DQ2 and DQ8 are very unlikely to have CD.
Non-HLA genes have a higher genetic contribution towards CD compared to HLA genes; however, the predisposition depends on a number of genes, each of which adds a minor contribution to disease development. This small effect size compounded with genetic heterogeneity between populations, has made non-HLA coeliac disease predisposing genes very difficult to identify and reproduce.
The CELIAC2 locus has been identified on chromosome 5, delimited by the genetic markers D5S410 and D5S402 within region 5q31-q33. This marker has been confirmed as a disease locus for CD in the recent European Cluster study on CD12. Though a number of inflammatory related genes exist in this area, the positive identification of the actual gene involved has remained very elusive.
The CELIAC3, corresponds to the CTLA4 region on chromosome 2. This region contains three genes, the CTLA4 gene, CD28 and ICOS, whose products are involved in the activation and control of the T-Cells. CTLA4 and CD28 cellular membrane proteins compete for the Antigen Presenting Cell (APC) membrane protein, CD80/86. If the latter product is missing, the T-cell undergoes apoptosis, whilst its interaction with CD28 induces T-cell proliferation and differentiation and its interaction with CTLA4, produces T-cell growth arrest. Thus, it was postulated that changes in the molecular structure of either of these two proteins could result in over-stimulation of the T-cell with the resultant intestinal damage typical of coeliac disease. In actual fact, single nucleotide polymorphisms (SNPs) within this group have been associated with the condition whilst in others, including the local coeliac population the same SNPs have been found to be protective.
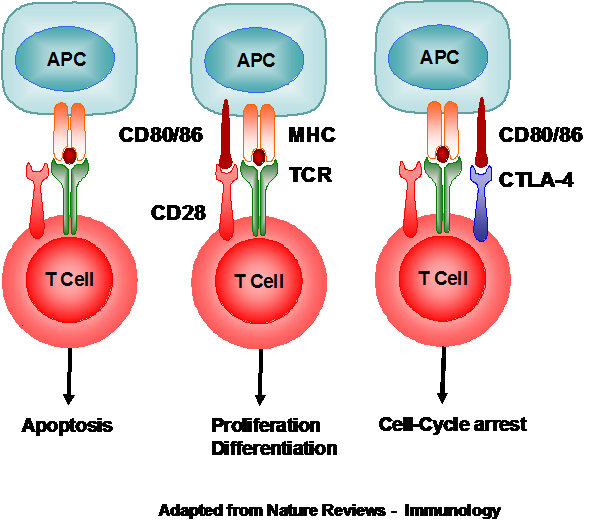
Figure 1. Interaction between the APC, T-cell, CD28, and CTLA-4.
CELIAC4 locus is located on chromosome 19p13.1. Studies on the CELIAC4 locus revealed a significant and replicable association to a common variant located in intron 28 of the gene myosin IXB (MYO9B). A defect in MYO9B may be a factor involved in the early mucosal events preceding the inflammatory response. A genetic variant in the 3-prime part of MYO9B may interrupt the tight junction gate, consequently the immunogenic gluten peptides can enter the deeper mucosal layer more easily.
In addition to the above, a number of genes have been associated with CD, in most cases following the genome-wide association study. Locally studies have been undertaken utilising a genome-wide linkage analysis approach on large families with multiple affected members. Though this work is still in its infancy, a novel mutation on the gene encoding the CD44 and another mutation for the gene encoding the CD59 cell surface proteins were identified.
As a conclusion, it appears that the genetic predisposition to CD depends on a single gene with a large effect (HLA-DQ2/DQ8), with a large group of other genes affecting the various aspects of innate and adaptive immunity and intestinal permeability.
References
1. Cooke W.T. and Holmes G.K.T. (1984). Coeliac disease. London: Churchill Livingstone.
2. Gee S. (1888). On the coeliac affection. St Bartholomew’s Hospital Journal 24:17–20.
3. Dicke W.K. (1950). Coeliakie. PhD thesis. University of Utrecht: the Netherlands.
4. Shuster, S., Watson, A.J. and Marks, J. (1968). Coeliac syndrome in dermatitis herpetiformis. Lancet May 25; 1: 1101-6
5. Marsh M.N. (1981). Studies of intestinal lymphoid tissue: the cytology and electron microscopy of gluten-sensitive enteropathy, with particular reference to its immunopathology. Scandinavian Journal of Gastroenterology Supplement ;70:87-106.
6. Van de W.Y., Kooy Y., van Veelen P., Vader W., Koning F. and Pena S. (2000). Coeliac disease: it takes three to tango! Gut 46(5):734-7.
7. Grodzinsky E., Franzen L., Hed J. and Strom M. (1992). High prevalence of celiac disease in healthy adults revealed by antigliadin antibodies. Annals of Allergy 69:66-70.
8. Fasano A. (1996). Where have all the American celiacs gone? Acta Paediatrics Supplement. May;412:20-4.
9. Catassi C., Fabiani E., Ratsch I.M., Coppa G.V., Giorgi P.L. and Pierdomenico
10. R. (1996). The coeliac iceberg in Italy. A multicentre antigliadin antibodies screening for coeliac disease in school-age subjects. Acta Paediatrics 412(Suppl):29-35
11. Lohi, S., Mustalahti, K., Kaukinen, K., Laurila, K., Collin, P., Rissanen, H., Lohi, O., Bravi, E., Gasparin, M., Reunanen, A. and Maki, M. (2009) Increasing prevalence of coeliac disease over time. Alimentary Pharmacology & Therapeutics 26 (9) 1217 – 1225
12. Babron M.C., Nilsson S., Adamovic S., Naluai A.T., Wahlström J., Ascher H., Ciclitira P.J., Sollid L.M., Partanen J., Greco L., Clerget-Darpoux F. European Genetics Cluster on Coeliac Disease. (2003) Meta and pooled analysis of European coeliac disease data. Eur J Hum Genet. 2003 Nov;11(11):828-34.