
Sickle Cell disease
Dr Laura Grech and Prof. Joseph Borg
Introduction
Sickle cell disease (SCD) is the most common monogenic disorder and its prevalence is high throughout large areas in sub-Saharan Africa, the Mediterranean basin, the Middle East and India because of the remarkable level of protection that the sickle cell trait provides against malaria.1 This stems from a selection pressure, in fact high frequencies of the variant gene correspond to areas of high malarial transmission. It is estimated that approximately 300,000 babies per year are born with sickle cell anaemia.2
SCD is an inherited haemoglobinopathy caused by a single amino acid substitution at the sixth residue of the beta (β)-globin subunit (p.Glu6Val) resulting in the production of the characteristic haemoglobin S (HbS).3,4 Under deoxygenation conditions (when Hb is not bound to oxygen), the haemoglobin tetramers, that include the two-mutant sickle β-globin subunits (HbS), can polymerize causing the erythrocyte to undertake a crescent or sickle cell shape (figure 1). These sickle cell shaped RBCs are rigid and dysfunctional, and also play a central role in acute and chronic clinical manifestations of SCD.
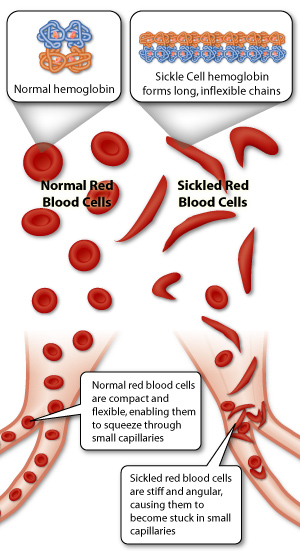
Figure 1: Demonstration of how sickle shaped RBCs give rise to vaso-occlusion events.5
The increased adhesiveness of the sickle cells causes microvascular obstructions in capillaries giving rise to blockage of blood flow with ischaemic/reperfusion injury.6
Inheritance of SCD
SCD is inherited as an autosomal codominant trait4. The most clinically relevant genotype is when the HbS mutation is inherited in homozygous fashion, that is when an individual inherits both the HbS alleles. This is also known as homozygous haemoglobin SS disease (HbSS). The HbS mutation can also be inherited in heterozygous form with other mutations. The most common double heterozygous mutation is an S mutation coupled with a thalassaemia or with haemoglobin C (HbC).7 These forms have the same characteristic clinical feature as HbSS but severity can vary, in fact when inherited with HbC the severity is milder than when inherited with thalassaemia. The phenotypic variability can also be due to an increase in foetal haemoglobin (HbF) levels. Subjects suffering from SCD with high HbF show a milder phenotype. On the other hand, if a person is heterozygous for the HbS allele the individual is said to be suffering from sickle cell trait (HbAS) and it is generally clinically benign.8
Pathophysiology of homozygous SCD
SCD is a multisystem disorder that affects nearly every organ in the body as shown in figure 2. Vaso-occlusion, haemolytic anaemia and vasculopathy are the hallmark of SCD while hypercoagulability and inflammation are also involved in organ damage.
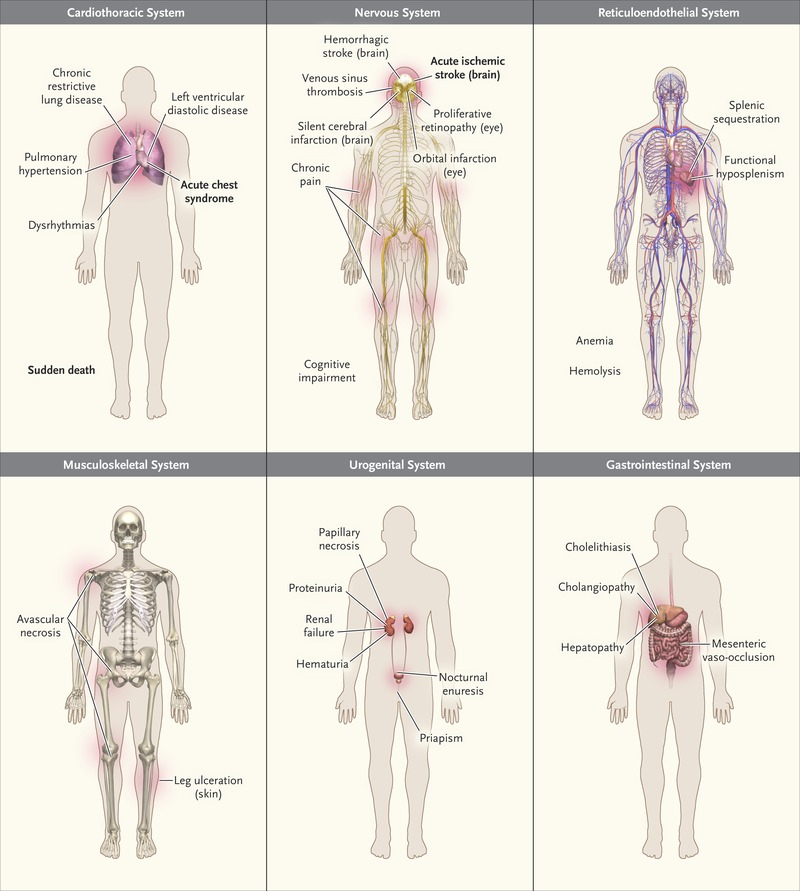
Figure 2: Common Clinical Complications of SCD.1
In SCD the essential pathophysiological occurrence is the HbS polymerization which gives rise to:
- Changes in Erythrocyte morphology – When compared to HbA (normal haemoglobin) the HbS has reduced oxygen affinity which aggravates HbS polymerization.9 HbS polymerization correlates exponentially with the concentration of HbS within the erythrocyte. As the polymer fibres extend, the erythrocytes are deformed and this interferes with their flexibility and flow properties giving rise to vaso-occlusion.10
- Altered erythrocyte membrane – HbS polymerization directly or indirectly alters the typical lipid bilayer and proteins of the erythrocyte membrane. This leads to early erythrocyte apoptosis due to reduced cellular hydration, increased haemolysis and abnormal interactions with other blood cells.11
- Haemolysis – Sickle erythrocytes are highly unstable with a lifespan that is reduced by >75%. Principally, haemolysis occurs via extravascular phagocytosis by macrophages, but about one-third occurs through intravascular haemolysis.12
- Oxidative stress – Haemolysis gives rise to oxidative stress which enhance HbS auto-oxidation contributing to the damage of the cell membrane, premature erythrocyte ageing and haemolysis.13
- Free plasma Hb and haem – Extracellular haemoglobin and haem in plasma promotes oxidative stress of blood vessels and blood cells. Superoxide is produced by the continuous auto-oxidation of extracellular haemoglobin. The superoxide dismutases into hydrogen peroxide which promotes further vasoconstriction.13
- Innate immune system activation – Neutrophils are activated by haem to release DNA as neutrophil extracellular traps which increase platelet activation and thrombosis and promote pulmonary vaso-occlusion.14
Clinical features of Homozygous SCD
In homozygous SCD clinical features are that of severe haemolytic anaemia punctuated by crises. The clinical expression of homozygous SCD is very variable; some patients develop severe crises even as infants and may die in early childhood or as young adults. Crises may be:
- Painful Vaso-occlusive Crises
In early infancy, childhood and adulthood patients with SCD may experience intense pain which usually accounts for the majority of hospitalisations. This pain which is described as one of the most unbearable forms of pain that affects human beings is caused by factors such as infection, acidosis, dehydration or deoxygenation. Pain occurs due to stimulation of nociceptive nerve fibres caused by microvascular occlusion. The microcirculation is obstructed by sickle-shaped RBCs which restrict the flow of the blood to the organ and results in ischaemia, oedema, pain, necrosis and organ damage.15 Infarcts can occur in a variety of organs such as the bone, the lungs and the spleen but the most serious vaso-occlusive crises are those that occur in the brain or spinal cord. In children the ‘hand-foot’ syndrome (painful dactylitis caused by infarcts of the small bones) is frequently the first presentation of the disease and may lead to digits of varying lengths. Resolution of pain is unpredictable. Acute pain might lead to chronic pain.2
- Visceral Sequestration Crises
The visceral sequestration crises are caused by sickling within organs and pooling of blood, often with a severe exacerbation of anaemia. After puberty, the most common cause of death is acute sickle chest syndrome which presents with dyspnoea, falling arterial PO2, chest pain and pulmonary infiltrates on chest X-ray. Splenic sequestration is typically seen in infants; these present with an enlarged spleen, decrease in haemoglobin and abdominal pain. Patients must be monitored at regular intervals and treatment is with transfusion. Since attacks tend to be recurrent, splenectomy is often needed.16
- Aplastic Crises
In SCD, aplastic crises can occur either as a result of infection with parvovirus or from folate deficiency. The virus affects erythropoiesis by invading progenitors of RBCs in the bone marrow and destroying them, thus preventing new RBCs from being made. In SCD the life span of RBCs is reduced to about 10–20 days. This results in a sudden fall in haemoglobin which usually requires transfusion.17
Other clinical features include ulcers of the lower legs as a result of vascular stasis and local ischaemia. In infancy and early childhood, the spleen is enlarged but as a result of infarcts, later on in life is often reduced in size. Other clinical complications include pulmonary hypertension, retinopathy, priapism and chronic damage to the liver.
Diagnosis of Homozygous SCD
Diagnosis of SCD involves the combination of blood counts and quantitative studies of haemoglobin fractions.
Blood Counts and Blood Films
In SCD the blood count and the erythrocyte indices depend on the presence or absence of confounding conditions such as iron or folic acid deficiency and coincidence of beta or alpha thalassaemia. In patients homozygous for SCD the haemoglobin is usually 6-9g/dL while if SCD is inherited together with alpha thalassaemia or beta thalassaemia the haemoglobin will be between 8-10g/dL. Figure 3 shows a blood picture with sickle cells, target cells, Howell-Jolly bodies (due to splenic atrophy), nucleated RBCs and polychromatophilic cells.
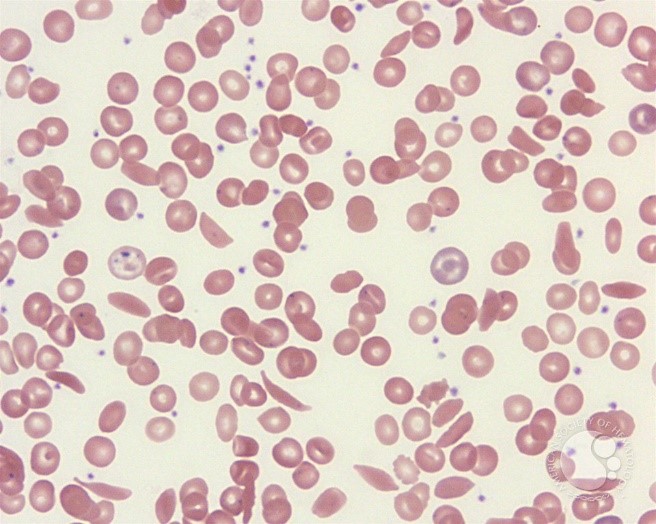
Figure 3: Peripheral smear from a patient with sickle cell disease illustrates the spectrum of RBC findings in this disorder including sickle cells, polychromatophilic RBCs, target cells and Howell-Jolly bodies.18
Detection of HbS and Measurement of Haemoglobin Fractions
In SCD, from neonatal life through early adult life, there is a slow but continual fall in HbF, whereas an increase in HbA2 level is seen until ages 1-2 years. The haemoglobin fractions present at one year of age are sufficiently stable to be relied on for diagnosis. In untreated SCD, more than 80% of the hemolysate is always HbS, except in infancy when the γ to β globin gene switching is incomplete. HbS can be detected by isoelectric focusing, haemoglobin electrophoresis or high-performance liquid chromatography. Since sickling haemoglobins are insoluble and precipitate in high-molarity phosphate buffer when reduced with sodium dithionite they can be detected chemically. The definitive test for SCD is DNA-based methods for the detection of HbS.19
Treatment
The only treatments approved for SCD are hydroxyurea (hydroxycarbamide, HU). Hydroxyurea which is a ribonucleotide reductase inhibitor acts in a number of ways, by (i) increasing foetal haemoglobin, (ii) modulating endothelial activation, and (iii) help to reduce chronic inflammation by reducing neutrophil counts.8 RBC exchange transfusion is usually used when HU therapy is not well-tolerated or not effective, such as in pregnancy and in SCD patients with acute or recurrent cerebrovascular disease. This can lead to iron overload or alloimmunization.20 The only curative option is bone marrow transplantation but it is a risky and costly treatment.
An improved understanding of both the pathogenesis and the pathophysiology of sickle cell-related organ damage based on mouse models for SCD has allowed new therapeutic options to be identified. These include agents that target SCD vasculopathy and sickle cell endothelial adhesive events. On the other hand, while various clinical trials on lentiviral gene therapy in SCD are currently ongoing.
References
- Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med 2017;377(3):305.
- Piel FB, Tewari S, Brousse V, et al. Associations between environmental factors and hospital admissions for sickle cell disease. Haematologica 2017;102(4):666-675.
- Pauling L, Itano HA. Sickle cell anemia a molecular disease. Science 1949;110(2865):543-548.
- Neel JV. The inheritance of the sickling phenomenon, with particular reference to sickle cell disease. Blood 1951;6(5):389-412.
- https://step1.medbullets.com/hematology/107040/sickle-cell-anemia5. Access date: 18 Dec 2019.
- Ballas SK, Smith ED. Red blood cell changes during the evolution of the sickle cell painful crisis. Blood 1992;79(8):2154-2163.
- Higgs DR, Aldridge BE, Lamb J, et al. The interaction of alpha-thalassemia and homozygous sickle-cell disease. N Engl J Med;306(24):1441-1446.
- Pinto VM, Balocco M, Quintino S, et al. Sickle cell disease: a review for the internist. Intern Emerg Med 2019;14(7):1051-1064.
- Seakins M, Gibbs WN, Milner PF, et al. Erythrocyte Hb-S concentration. An important factor in the low oxygen affinity of blood in sickle cell anemia. J Clin Invest 1973;52(2):422-432.
- Connes P, Alexy T, Detterich J, et al. The role of blood rheology in sickle cell disease. Blood Rev 2016;30(2):111-118.
- Kuypers FA. Hemoglobin s polymerization and red cell membrane changes. Hematol Oncol Clin North Am 2014;28(2):155-179.
- Crosby WH. The metabolism of hemoglobin and bile pigment in hemolytic disease. Am J Med 1955;18(1):112-122.
- Alayash AI. Oxidative pathways in the sickle cell and beyond. Blood Cells Mol Dis 2018;70:78-86.
- Wang X, Mendelsohn L, Rogers H, et al. Heme-bound iron activates placenta growth factor in erythroid cells via erythroid Kruppel-like factor. Blood 2014;124(6):946-954.
- de Montalembert M. Management of children with sickle cell anemia: a collaborative work. Arch Pediatr 2002;9(11):1195-1201.
- Rezende PV, Viana MB, Murao M, et al. Acute splenic sequestration in a cohort of children with sickle cell anemia. J Pediatr (Rio J) 2009;85(2):163-169.
- Minhas PS, K Virdi J, Patel R. Double whammy- acute splenic sequestration crisis in patient with aplastic crisis due to acute parvovirus infection. J Community Hosp Intern Med Perspect 2017;7(3):194-195.
- https://imagebank.hematology.org/image/3958/sickle-cell-disease-ndash-rbc-morphology–1?type=upload. Access date: 18 Dec 2019.
- Hoffbrand AV,Moss PAH, Pettit JE: Genetics Disorders of Haemoglobin. In: Hoffbrand AV, Moss PAH, eds. Essential Haematology. Oxford, UK: Blackwell Publishing Ltd, 2006. 5th edition. p.85-92.
- Iughetti L, Bigi E, Venturelli D. Novel insights in the management of sickle cell disease in childhood. World J Clin Pediatr 2016;5(1):25-34.